|
Eva Fariña. Madrid
El Dr. Luis Tercedor, jefe de Sección de Cardiología de la Unidad de Arritmias del Hospital Universitario Virgen de las Nieves de Granada, ha explicado para Publicación Médica de Cardiología las novedades relativas a las canalopatías, y más concretamente al Síndrome de QT largo, que podría afectar, aproximadamente, a uno de cada 2.500 bebés nacidos vivos, si bien no todos los portadores desarrollan el problema durante su vida. Cuando esto sí ocurre, el paciente puede sufrir pérdida del conocimiento, si bien la peor consecuencia es la parada cardiaca por fibrilación ventricular. Este especialista ha indicado que “es imposible detectar todos los casos, pero sí es importante identificar a los pacientes que ya están mostrando síntomas, sobre todo la pérdida de conocimiento”. En su opinión, “es fundamental la detección precoz para evitar que degenere en un problema más serio”.

El Dr. Luis Tercedor es jefe de la Unidad de Arritmias
del Hospital Virgen de las Nieves de Granada.
Foto: Sociedad Española de Cardiología.
|
En el electrocardiograma de superficie, el QT es el intervalo que mide el inicio del QRS hasta el final de la onda T. Las canalopatías incluyen el síndrome de QT largo y el síndrome de Brugada, entre otras enfermedades de los canales iónicos. “Dichos canales son estructuras de las membranas celulares, con permeabilidad selectiva a los electrolitos, principalmente sodio y potasio. El tráfico de estos electrolitos es fundamental en la generación del potencial de acción, que es lo que genera la actividad eléctrica del corazón. Cuando se alteran estos canales iónicos, se produce la canalopatía. Generalmente se trata de una enfermedad eléctrica, puesto que en el corazón no se observa ninguna alteración estructural”, ha explicado este especialista.
“Estos canales, que están en diversas partes del organismo además de en el corazón, son estructuras proteicas fundamentales para la vida. Muchas veces las funciones de los distintos canales se superponen un poco. El defecto de un canal generalmente es debido a anomalías genéticas, es decir, son mutaciones puntuales en un solo gen. En algunas ocasiones, el déficit que se origina en este canal no es muy importante; es decir, disminuye su función, pero en un porcentaje pequeño, y puede hacer que los otros canales cumplimenten su función. Esto provoca que la enfermedad en situación basal sea difícil de ver. Normalmente se identifica por el electrocardiograma, que es donde se detecta la alteración eléctrica, pero el problema de las canalopatías es que esa situación basal de la persona puede que esté compensado ese déficit del canal, y el electro sea prácticamente normal. Esto se conoce como 'penetrancia incompleta'”.
“Entre los desencadenantes están las situaciones de estrés y los ruidos intensos; también el periodo postparto es una época de riesgo; y, a veces, la administración de algunos fármacos. Hay muchos medicamentos que pueden bloquear los canales, no solo arrítmicos, sino de los que se utilizan habitualmente, como antihistamínicos o antibióticos. En algunas ocasiones, la enfermedad se pone de manifiesto con alguno de estos desencadenantes, y el problema es que cuando aparece, si no se ha diagnosticado antes, puede debutar con alguna arritmia grave. Las canalopatías cardiacas, en concreto el síndrome de QT largo, produce un tipo de taquicardia ventricular polimórfica, la ‘torsión de punta’. Generalmente son episodios que se autolimitan y pueden producir una pérdida de conciencia con recuperación espontánea del paciente que lo sufre, pero, a veces, puede que se mantengan y degeneren en un paro cardiaco por fibrilación ventricular. Es decir, la presentación puede ser grave”.
 |
El QT largo, según ha informado el Dr. Tercedor, “tiene un tratamiento farmacológico muy eficaz, que previene los episodios graves, incluso la muerte súbita”. “El QT largo se trata con betabloqueantes, que son fármacos de uso cotidiano en la Medicina y en la Cardiología, y son medicamentos muy seguros y muy eficaces”. Es crucial hacer el diagnóstico pronto, porque cuando el paciente inicia el tratamiento betabloqueante se reduce de manera muy eficaz el riesgo de sufrir eventos cardiovasculares graves. “El problema al que nos enfrentamos es que el diagnóstico por electro no funciona al cien por cien. En el caso del QT largo tenemos la enorme ayuda de los test genéticos, que son muy sensibles y detectan el 70 por ciento de los casos. Un gran porcentaje de pacientes se va a encontrar una mutación patogénica, si bien en el 30 por ciento de los casos no se va a detectar ninguna mutación, a pesar de ser portadores de la enfermedad”.
Tratamiento con betabloqueantes y posibilidad de implantar un desfibrilador
En cuanto al tratamiento, el Dr. Tercedor ha explicado que “para aquellos pacientes que no hayan sufrido un paro cardiaco, el tratamiento inicial es con betabloqueantes, que son muy eficaces”. “Es crucial saber que bajo ningún concepto los pacientes deben dejar de tomar estos fármacos, son tan necesarios como la insulina para los diabéticos. Además, es fundamental que estos pacientes tengan un listado con todos los fármacos que pueden actuar bloqueando estos canales iónicos que están afectados, y desencadenar una arritmia. La lista de fármacos es muy extensa, y se puede consultar en internet. Muchas veces ni siquiera está especificado en la ficha técnica del medicamento el riesgo de que pueda desencadenar o agravar un síndrome de QT largo”.
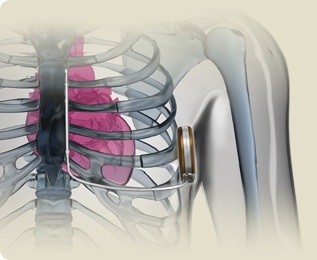
Los pacientes que ya presentan un paro cardiaco, “además del tratamiento farmacológico, también necesitan un desfibrilador implantable”, según ha dicho el especialista del Hospital Virgen de las Nieves: “Este dispositivo no previene que aparezca la arritmia, pero es muy eficaz en el caso de que se presente un paro cardiaco por fibrilación ventricular, ya que puede recuperar al paciente y evitar las consecuencias”. “Antes de que se produzca un paro cardiaco, también es aconsejable implantar un desfibrilador a aquellos pacientes que están tomando los betabloqueantes y no son eficaces, porque recurren las arritmias o los síncopes. Cuando concurren una serie de marcadores electrocardiográficos, como el QT muy prolongado asociado a sordera congénita (Síndrome de Jervell-Lange-Nielsen), la situación de riesgo es alta, por lo que, además del tratamiento con betabloqueantes, también se puede indicar el desfibrilador implantable”.
Según el Dr. Luis Tercedor, también puede ocurrir que portadores de una mutación para Síndrome de QT largo nunca desarrollen el problema. “Los portadores de esta canalopatía que nunca en su vida se ven expuestos a situaciones de estrés o no toman fármacos que desencadenan el síndrome puede que nunca lleguen a manifestar la enfermedad. De hecho, esto es relativamente frecuente en los familiares de pacientes que han tenido un problema de arritmias, ya que muchos son portadores silentes o silenciosos de la anomalía genética. En las canalopatías, a diferencia de lo que ocurre en las miocardiopatías familiares, el antecedente familiar de paro cardiaco no es un marcador fiable de riesgo de muerte súbita en los familiares afectos”.
|